Systemic lupus erythematosus (SLE) is a chronic, systemic autoimmune disease, particularly prevalent in women, probably with a genetic predisposition with “triggering” from contact from an environmental stimulus, resulting in the production of pathogenic autoantibodies and immune complexes which produce the pathologic features of the disease.
SLE has protean manifestations and is often difficult to diagnose in its early stages. The diagnosis can be definitively established when at least four of the eleven American College of Rheumatology criteria are met, either serially or simultaneously. Although ocular inflammation may be a manifestation of SLE—and may even be the initial clinically apparent feature—the ocular symptoms and signs are not included among the eleven criteria; we believe this is an oversight and believe, further, that inclusion of ocular inflammation among the diagnostic criteria for SLE would enable earlier establishment of the diagnosis and earlier therapeutic intervention in some instances.
Corneal manifestations of SLE are confined primarily to ocular surface epitheliopathy secondary to keratoconjunctivitis sicca, and stromal keratitis (rare), particularly peripheral and segmental. In our 47 patients with SLE which we have analyzed, 16 had corneal complications, 62.5% secondary to keratoconjunctivitis sicca, and the rest secondary to the lupus disease activity itself.
Episcleritis or scleritis may also occur as a manifestation of SLE, and scleritis is a reasonably accurate guide to the presence of significant systemic activity in the SLE patient. Scleritis typically resolves only with adequate control of the disease activity and will not respond to topical therapy. SLE was present in 1 of 94 patients with episcleritis and in 7 of 172 patients with scleritis in our reported cohort. Systemic nonsteroidal anti-inflammatory drug therapy was necessary to treat SLE-associated episcleritis, unlike the usual situation in patients with idiopathic episcleritis.
Retinal involvement is the most common ocular manifestation of SLE after keratoconjunctivitis sicca. Additionally, the presence of active SLE retinal vasculopathy is an extremely accurate guide to the presence of systemic disease activity, occult or overt. Life-table survival estimates have shown decreased survival in patients with SLE retinopathy, compared to SLE patients without retinopathy. Lupus retinopathy should alert the clinician to the likelihood of ocular and systemic vasculitis involvement and may warrant aggressive systemic therapy. Retinal vasculitis appears early in the development of lupus retinopathy and is evident on fluorescein angiography. Subclinical macular edema, intraretinal hemorrhages, and cotton-wool spots may follow; these latter changes may be indistinguishable from hypertensive retinopathy, making it difficult for the clinician to determine whether the retinal lesions are secondary to hypertension or to SLE immune-complex vasculitis. Aggressive therapy for hypertension and SLE systemic disease activity is associated with a dramatic decrease in retinal lesions in such patients and improves the patient’s overall prognosis. The appearance of SLE retinopathy is associated with central nervous system involvement. Additionally, 5% to 10% of patients with SLE retinopathy will develop large vessel disease such as retinal artery occlusion and retinal vein occlusion in the presence of anti-phospholipid antibodies (anticardiolipin antibodies, lupus anticoagulant, and anti–β2 glycoprotein I antibodies). Corticosteroid and Heparinization may be critical in treatment of this form of lupus retinopathy.
Finally, choroidopathy, choroidal vascular occlusion, and choroidal vasculitis may occur in patients with SLE, resulting in serous retinal detachment.
 |
|
|
|
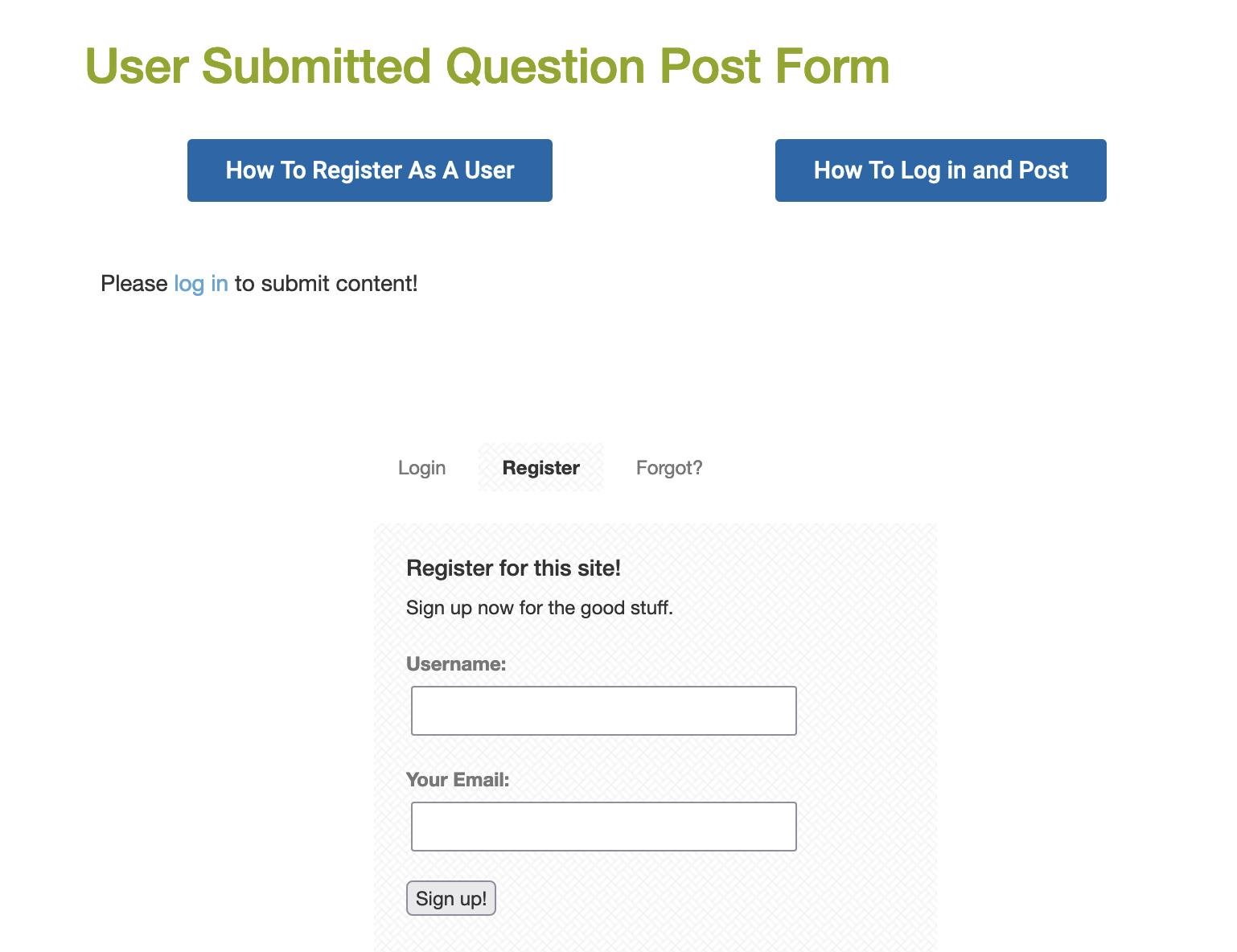
Click on the Register link
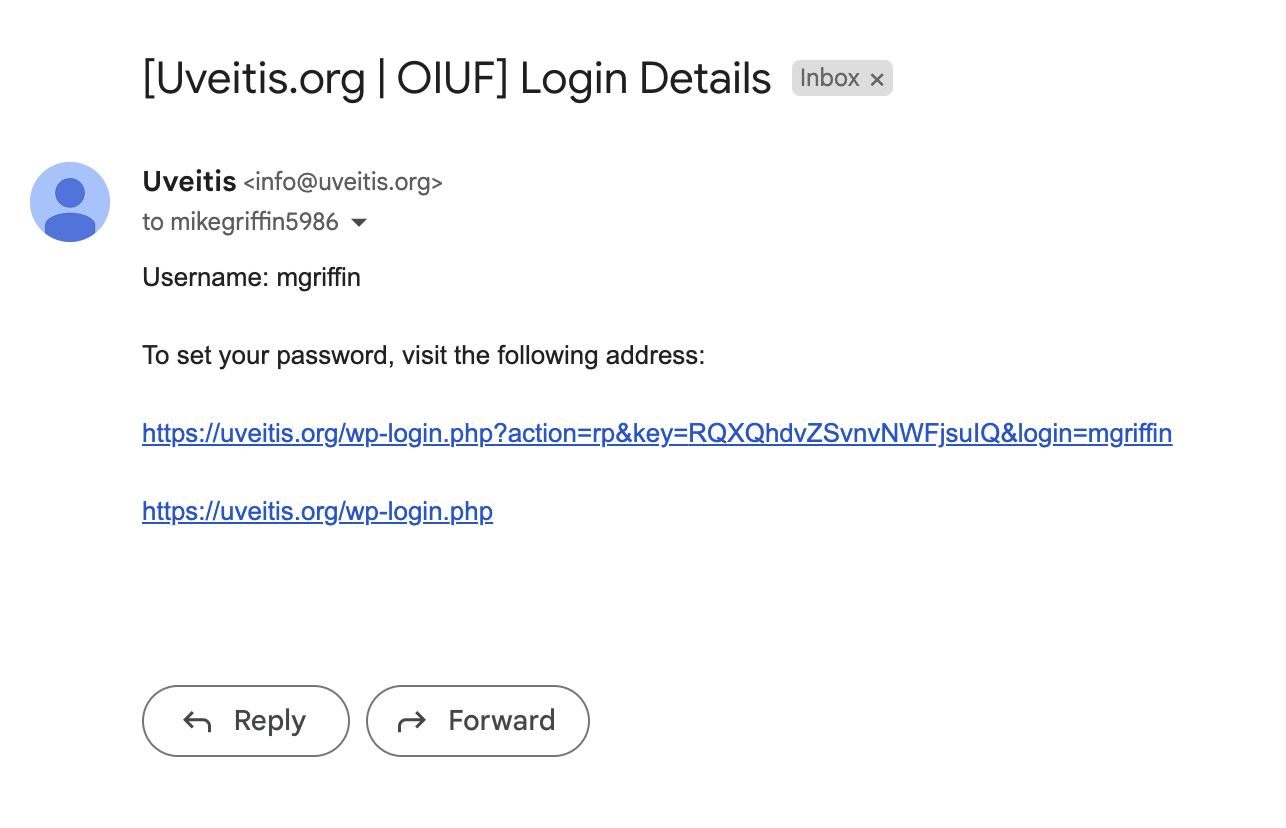
In the email you receive, click on the change password link.
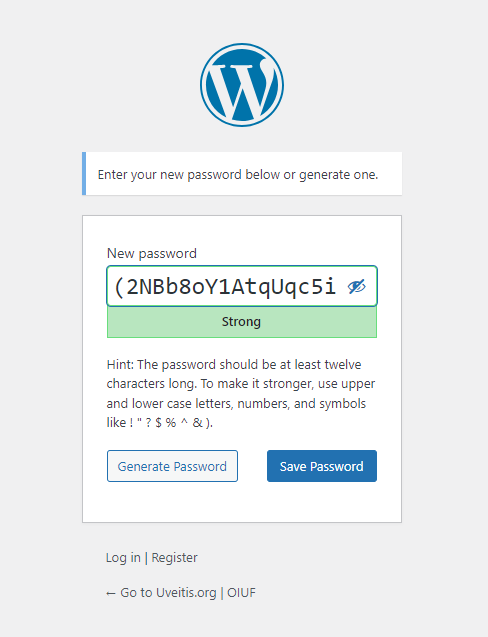
Change your password