Two of the postdoctoral ocular immunology fellows , along with a biostatistician have analyzed the records of 100 patients (138 eyes) with episcleritis evaluated by us over the past decade or more. Data were extracted from the medical records and placed into a customized database software package to determine the frequency and type of systemic disease eventually discovered to be associated with each type diffuse vs. nodular of episcleritis.
The mean age at presentation to our service was 43 years (range 18-76), with females predominating (female/male = 7/3). One-third of the patients had bilateral involvement. 16% had nodular episcleritis, and in 28% of the patients the episcleritis was recurrent. Associated systemic disease was found in 1/3 of the patients (36%). One-half of the patients had concurrent other ocular involvement, including uveitis (13%), glaucoma (5%), and keratitis (13%). In only 4 did the episcleritis progress to true scleritis. The most serious systemic disease discovered in the course of investigating these 100 patients was in a single patient with episcleritis who was discovered to have Granulomatosis with Polyangiitis (formerly called Wegener’s).
Most patients required no treatment except “supportive” care with iced artificial tears, but some have had at least a short course of oral nonsteroidal anti-inflammatory drug therapy (often non-prescription strength).
Conclusions: Although episcleritis is typically a benign and self-limited disease and therefore does not need topical steroid therapy, some patients do require treatment and we believe that the appropriate treatment is with low-dose oral nonsteroidal anti-inflammatory drug therapy. Additionally, since 1/3 of the patients may have associated occult systemic disease, an extremely careful review of systems history taking is appropriate, with appropriate laboratory testing based on the pertinent positives emerging from that survey.
 |
|
|
|
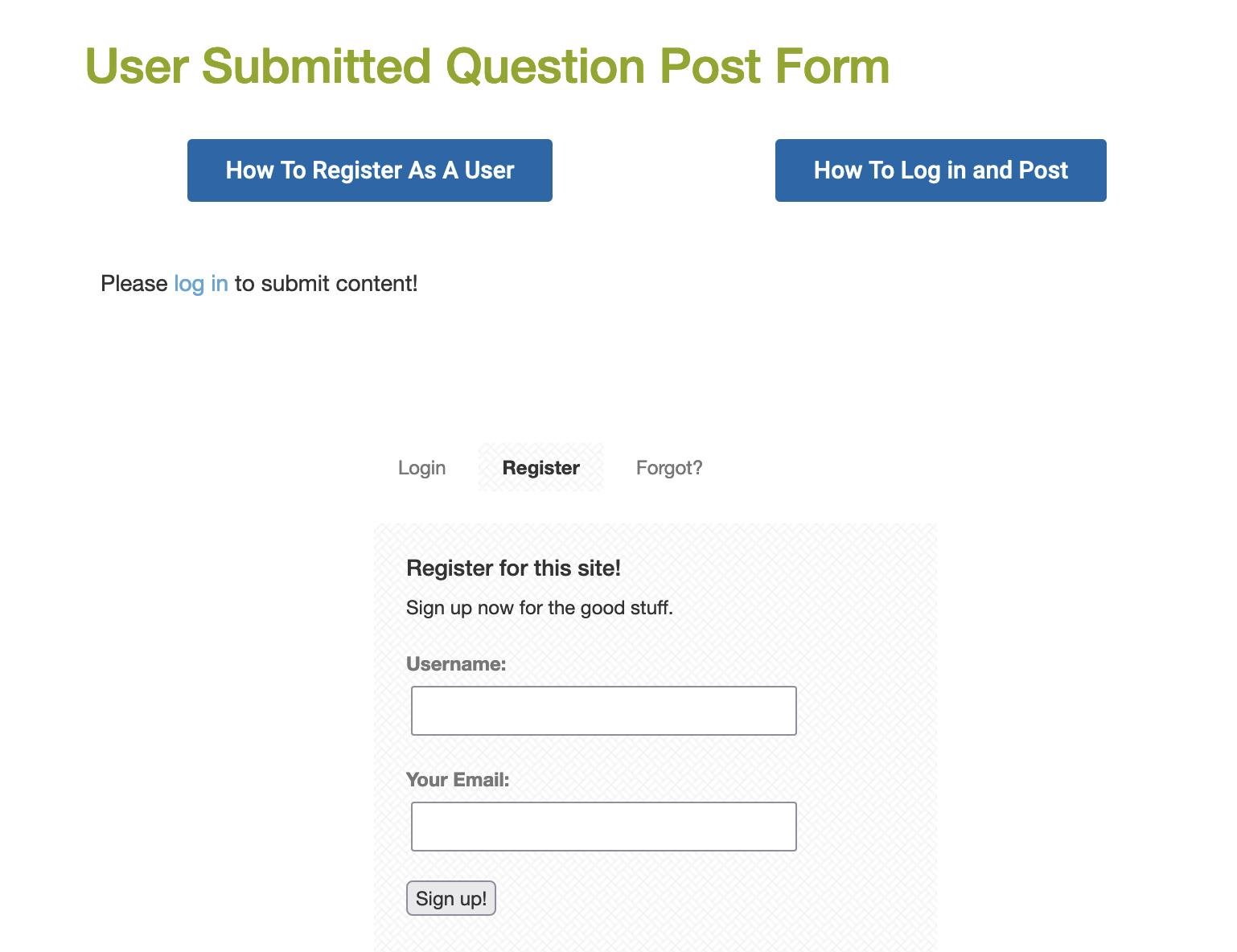
Click on the Register link
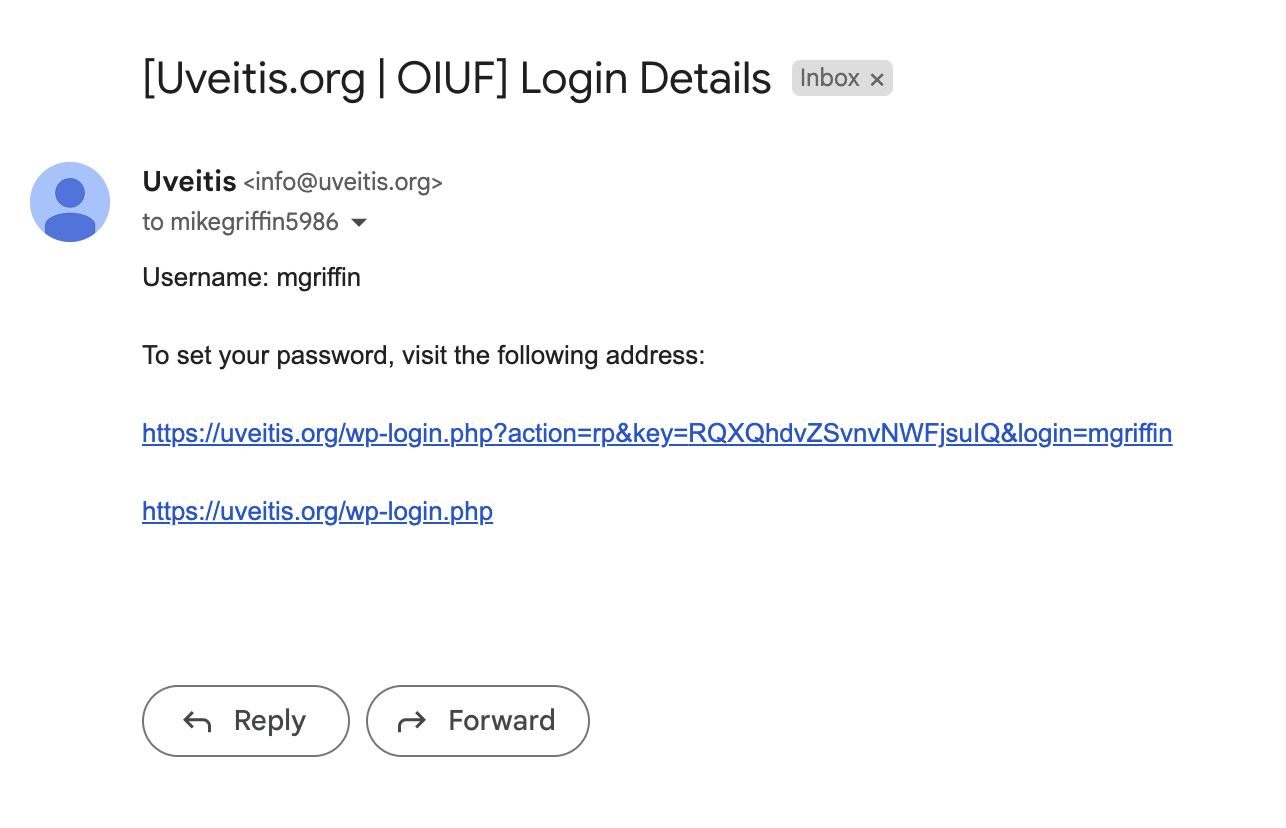
In the email you receive, click on the change password link.
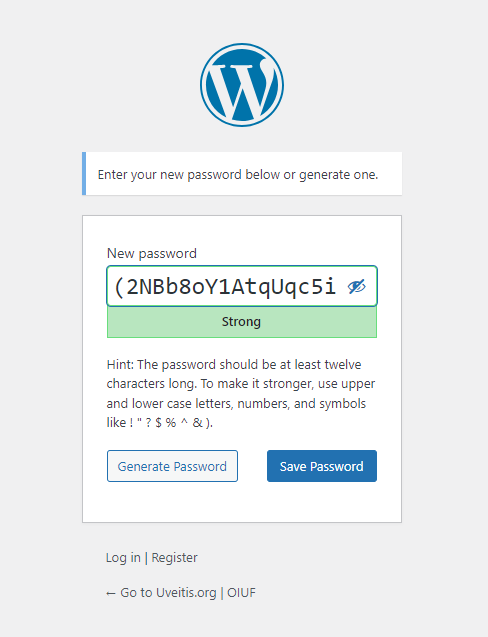
Change your password